
Thrust 1: Mapping Free-Energy Landscapes
Our proposed multi-scale framework will combine first-principles electronic structure calculations, atomistic molecular dynamics (MD) and Monte Carlo (MC) simulations, and coarse-grained (CG) models to simulate assembly at multiple resolutions. Each approach will provide predictions at its operational length and time scale, as well as information for parameterizing the next coarser model in the hierarchy of models. First, information from chemical synthesis and characterization (Thrust 2) will be parametrized by Arya and Paesani in order to build CG models that capture the distinctive features for each NC or protein building block. This will include features such as chemical composition, nanoscale dimensions obtained from electron or optical microscopy, polymer graft lengths, and the location of programmed binding sites. CG models built by Arya will then provide the effective interactions between nanoscale building blocks and will be used to analyze distance- and orientation-dependent free-energy trajectories associated with binding (i.e., two-body assembly).
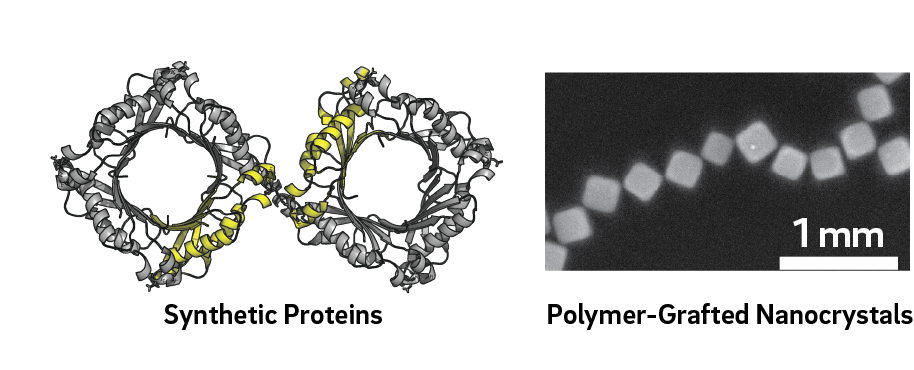
For example, polymer-grafted Ag NCs studied by Tao were translated into CG models by Arya with: (i) NCs treated as cubic lattices of Ag atoms, and (ii) grafted chains treated as CG bead-chains. Comparing the free energy of interaction for different NC orientations (e.g., face-face or edge-edge binding) allowed us to determine the most favorable orientation and equilibrium separation distance between two shaped NCs. This free-energy landscape provided direct feedback for Tao’s fabrication of plasmonic nanojunctions, where graft length was a synthetic “knob” for controlling optical resonance wavelength. In Thrust 1, Arya, Paesani, and Tezcan will work together to develop similar CG models of synthetic tetrameric proteins (RhuA and FolB) possessing four-fold symmetry that Tezcan has genetically programmed with corner binding sites. Input into the CG model will include: protein composition; protein shape/topology as determined from electron microscopy and crystallography; nature and location of binding sites (e.g., Cys residue located at position 15 in FolB); and expected number of binding sites per protein. Atomistic MD simulations will provide information on the conformations of proteins and linkers to help parametrize molecular-level information for the CG model. CG protein models will be used to identify equilibrium assembly structures that result in the formation of open- or closed-pore structures.
Our multi-resolution framework will connect mesoscopic CG models with atomic- and molecular-level details elucidated at the quantum mechanical (QM) level, which will enable us to investigate previously inaccessible phenomena associated with assembly. For example, solvent chemistry and dynamics, which are known to dictate the assembly pathway of biomolecules as shown by Paesani, are minimally explored in models of larger colloidal systems. Basing all subsequent levels of modelling on QM allows our framework to be robust, predictive, and sufficiently accurate to describe local electrostatic environments (e.g., solvent interaction, adsorbates) for our nanoscale building blocks. In demonstration of this overall concept, Paesani modelled C98RhuA proteins synthesized by Tezcan that are “hinged” via intermolecular disulfide bonds (located by a Cys residue at position 98) and rotate in a coherent fashion to form an elliptical pore. Paesani used all-atom umbrella sampling to evaluate the energy landscape of C98RhuA proteins in the presence of >100,000 water molecules. The calculated energy profiles of C98RhuA interactions as a function of pore ellipticity showed that equilibrium conformations for a single pore structure were dictated by solvent entropy. In Thrust 1, Paesani will work with Arya and Tao to carry out QM calculations that provide accurate predictions of polymer graft structure and NC energetics in the presence of solvent molecules (validated by Pascal via simulated spectroscopy in Thrust 4). The multibody effects associated with these molecules (e.g., interactions with polymer grafts, adsorption to metal surface) will be taken into account at the CG level by using the data-driven many-body expansion approach developed by Paesani.
In situ electron microscopy will be used to validate the free-energy trajectories developed by Arya, Paesani, Tao, and Tezcan. Liquid cell transmission electron microscopy (LC-TEM) will be used to directly image two-body binding events (~10 seconds). We will develop LC-TEM protocols to study NC and protein binding on UCSD’s recently acquired Talos F200X. Tao will also develop a new in situ technique called polymer-assisted scanning electron microscopy (PA-SEM) which is capable of direct-imaging particle interactions within a larger field of view (i.e., over many particles) and over longer time scales (~minutes) compared to LC-TEM. In the proposed PA-SEM technique, Tao will adapt graphene LC-TEM cells to encapsulate polymer thin-films that can then be imaged at temperatures above their softening point (for polystyrene MW=10K, T~80°C). The polymer serves as a high viscosity solvent and has been observed to telescope the dynamics of NC assembly over tens of minutes, including diffusion and collision events.
For example, Arya’s calculations predict that the transition between face- to edge-linked NCs occurs through stepwise stick-and-slip type motions rather than a single smooth rotation. PA-SEM will be used to measure the timescale and trajectory for NC binding. Possible disagreement with the model will provide feedback for re-evaluation of our CG model for accuracy. Binding trajectories for a repertoire of building blocks will be compiled into a searchable database and used to ascertain free-energy trajectories via a data-driven route. This knowledge will then be made available to broadly inform assembly strategies for a wide range of materials systems.